Atomistic computer simulations are virtual experiments that complement (but not replace) real experiments. In increasingly many situations they can give an atom-scale understanding, a microscopic explanation of condensed phase electronic and chemical processes that is often difficult to obtain from real experiments. Yet, major limitations are the approximations necessary to solve the underlying equations of many-particle systems (particularly the electronic Schrodinger equation), and the time and length scales that are accessible to computer simulations. Our group carries out research (i) to further advance the predictive power of atom-scale computer simulation techniques and (ii) to raise their significance by developing accurate multi-scale models that bridge the gap between the atomistic and the experimentally relevant time and length scales. While much of the research being carried out is fundamental in nature, we have a keen interest to apply the methodologies developed to relevant problems in the energy and health sector.
A major current research focus is the computer simulation of charge transport processes in soft condensed phase systems such as organic semiconductors and proteins as well as at solid/liquid interfaces relevant for (photo)electrochemical applications.
Non-Adiabatic Dynamics in Organic Solar Cell Materials
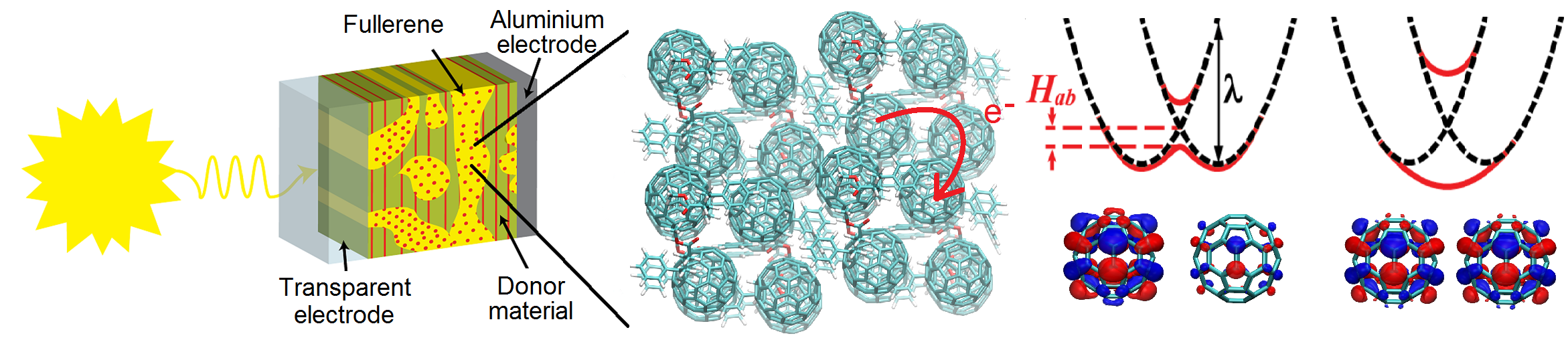
Charge transport (CT) in soft condensed matter is at the heart of many exciting and potentially revolutionising technologies ranging from organic photovoltaic cells to nanobioelectronic transistors. Tremendous progress has been made on these research frontiers over the last twenty years. Yet, our fundamental understanding of CT in organic and biological semiconductors that could rationalise experimental observations and guide further advances in the field is still very limited. These materials are characterised by strong, anharmonic thermal fluctuations and small energy barriers for CT, which renders standard theories such as band theory or activated electron hopping in many cases entirely inadequate (Chem. Rev. 2017). Over the last few years we have developed an efficient mixed quantum-classical non-adiabatic molecular dynamics simulation method termed Fragment Orbital-Based Surface Hopping (FOB-SH) that has opened the door for ground-breaking new insight into this problem (PCCP 2019). We found that in these materials electrons form flickering polarons, objects that are half way between waves and particles. They are delocalized over up to 10–20 molecules in the most conductive organic crystals and constantly change their shape and extension under the influence of the thermal motion of the atoms (Nature Commun. 2019, Adv. Theory. Simul. 20) Click on link in the column to the right to watch movies of flickering polarons in organic crystals. We are now exploring how flickering polarons can be made larger and more wave-like, which would further increase the conductivity of organic materials and enable new applications in flexible electronics. We also plan to extend the FOB-SH methodology to simulate the electronic processes in organic solar cells including exciton transport and dissociation, charge separation and recombination.
Electron Transfer Reactions in Proteins
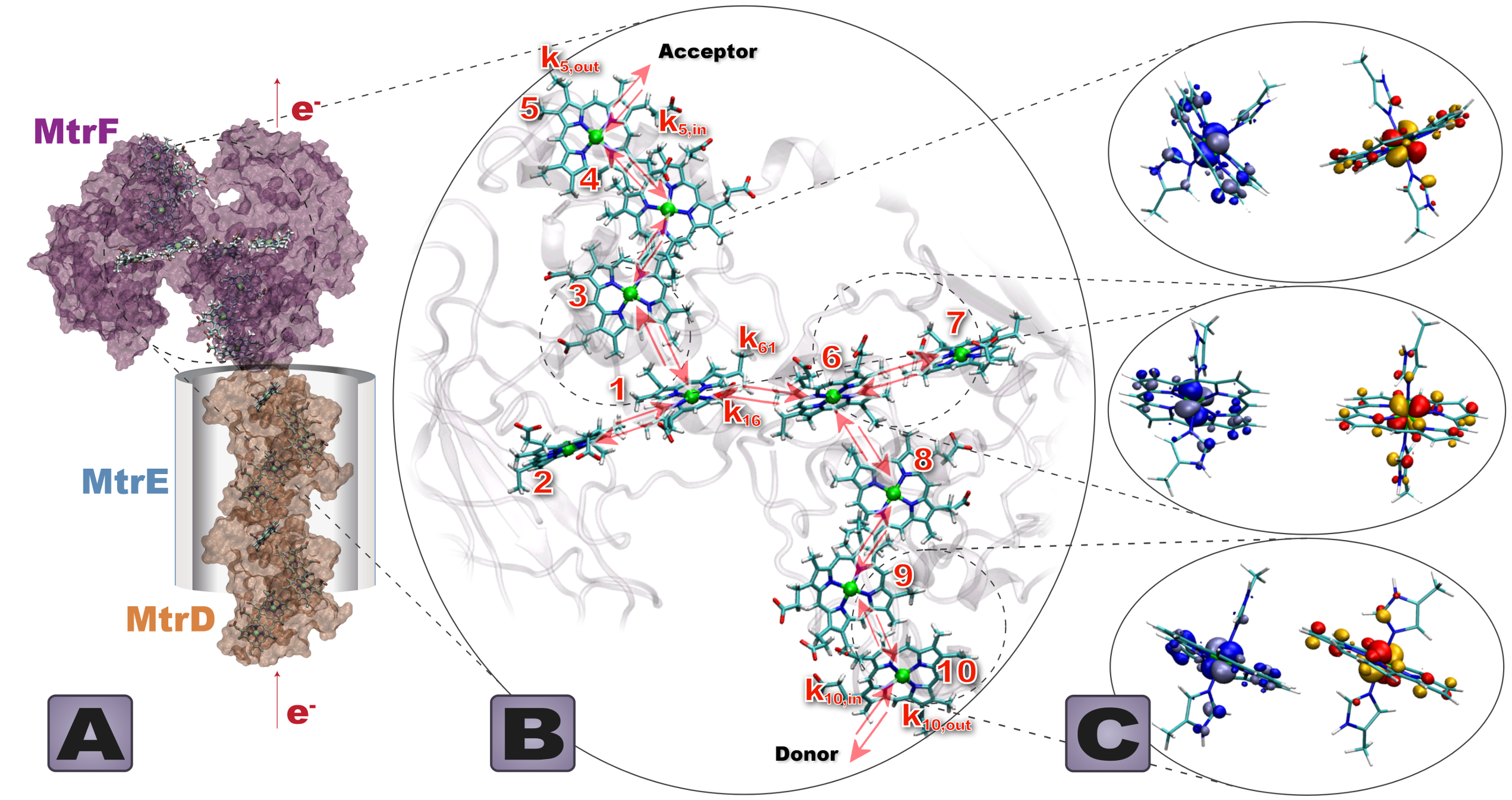
Certain microbes like Shewanella oneidensis are equipped with a fascinating biomolecular machinery that allows them to transport electrons from the inside of the cell across the bacterial cell membrane to extracellular space over distances exceeding 100 Angstroms. This way the microbe can reduce extracellular minerals like iron-oxide when oxygen is a limited resource, literally “breathing rocks” instead of oxygen. The biomolecules mediating the electron transfer to extracellular space are multi-heme proteins (see our review, JRSI 2015). They contain a large array of densely spaced, redox-active heme cofactors, which is why they are often referred to as “nano-wires”. We have recently calculated the thermodynamics and kinetics of ET in multi-heme proteins MtrC and MtrF (PNAS 2019) as well as in the large multiheme protein complex MtrCAB and the polymeric OmcS (JPCL20) using quantum mechanical and classical molecular dynamics simulation methods. Some of our predictions could recently be confirmed experimentally using ultrafast transient absorption spectroscopy (JACS 2019). We currently aim at obtaining molecular-level insight into the transport mechanism and electron flux that these proteins support when sandwiched between two electrodes in protein mono-layer junctions (JPCL20) and in single-molecule electrochemical scanning tunneling microscopy (EC-)STM junctions. This will inform their future use in semi-artificial photosynthesis and bioelectronic applications.
Oxide/Liquid Water Interfaces for Solar Fuels

Interfaces between transition metal oxides and liquid water have attracted research attention for quite a long time. In particular, the potential of photoelectrochemical (PEC) water splitting cells to provide carbon neutral H2 or fuels from CO2 (artificial photosynthesis) is in the focus of the research community. Our aim is to learn about the atomistic structure and the mechanism of charge transfer reactions at solid/liquid interfaces, such as e.g. the hematite/water system, using density-functional based molecular dynamics simulation. We are particularly interested in the microscopic structure (e.g. protonation states) of the interface (JACS 2017), the nature and transport properties of excess charge carriers in photoelectrodes (PCCP 2020), and the thermodynamics and kinetics of charge transfer across the solid/liquid interface (JPCL 2018) This knowledge may lead to atom-scale modifications that could improve photoanodes for solar water splitting.
Ligand Transport Within Proteins

The diffusion of small ligands within enzymes and their binding to enzyme active sites are ubiquitous molecular processes in biology. But how does a small molecule like H2, O2 or CO2 find its way through a dense and heterogeneous protein medium to the active site? Here we have developed a multiscale molecular simulation approach, where MD simulation and Markov state modelling are combined to shed light on the kinetics of ligand diffusion and binding (JACS 2011). For the proteins studied so far we have found that the ligands move within multiple hydrophobic cavities or `tunnels’ towards a central cavity from where the ligand makes the final transit to the catalytic site. The functional role of the protein can be compared to the one of a funnel guiding the small molecules towards the target (PNAS 2012, JACS 2013). We also extended our method to calculate sensitivity maps of the protein that identify residues (`hot spots’) that are expected to show the greatest affect on ligand diffusion when mutated (JCTC 2015). Such in-silico predictions could help protein engineers to optimize or control the kinetics of ligand, substrate or inhibitor diffusion in proteins as we have recently shown for FeFe hydrogenase (Nature Chem. 2017).