Atomistic simulation of electron transfer reactions in condensed phases has become a major task in our group. Such reactions essentially involve the transfer of an electron from a donor to an acceptor in a homogeneous or heterogeneous medium over length scales ranging from Angstroms to several nanometers. Electron transfer reactions form the basis of many energy-transforming processes in Nature (e.g. photosynthesis, respiration) and industry (e.g. solar cells and fuel cells). We are particularly interested to develop and devise simulation methods and protocols for the prediction of electron transfer rates and mechanism from first principles.
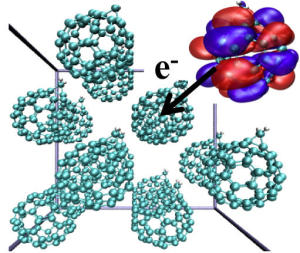
Crystal structure of a fullerene derivative. The orbital of an electron localized on one fullerene molecule is shown in blue and red isosurfaces and the electron transfer to a neighbouring fullerene molecule is indicated by an arrow. Fullerene-based materials are currently used as n-type semiconductors in organic solar cells, see picture below.
The application of modern density functional theory to electron transfer reactions is hampered by the approximate nature of the exchange-correlation functional. They tend to erroneously delocalize the excess electron, thus prohibiting an accurate modelling of charge transfer states. This deficiency termed electron self-interaction or delocalization error is intrinsic to GGA and hybrid density functionals unless special care is taken in their parametrization.
A method to circumvent this problem is constrained density functional theory (CDFT). In this approach an external potential is added to the Kohn-Sham equations preventing an excess electron or hole from wrongly delocalizing over donor and acceptor ions. The external potential is varied until a given constraint on the density, for instance a charge or spin constraint, is satisfied.
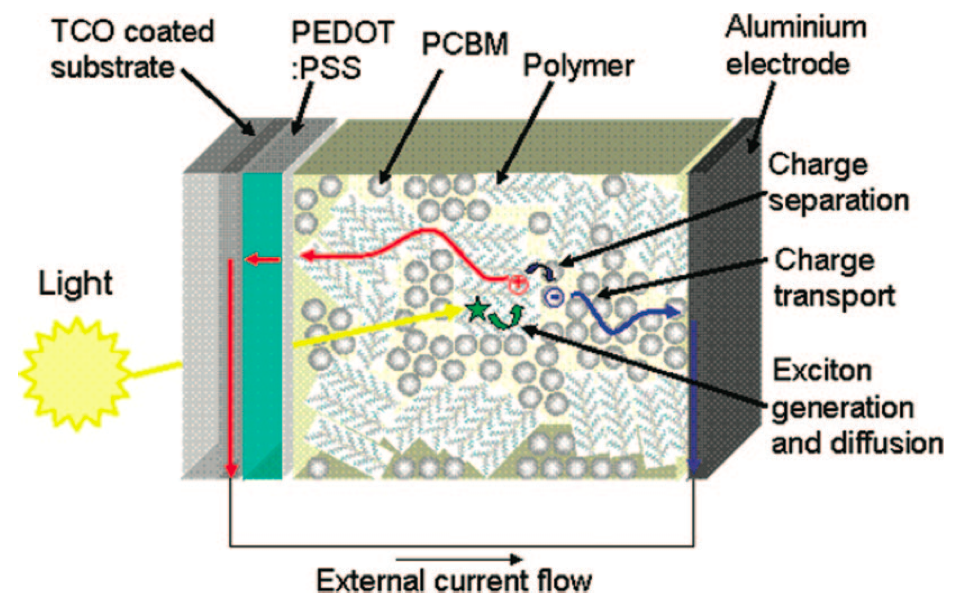
Negative charge transport, indicated by a blue arrow, is shown magnified in the picture above. Taken from Clarke et al., Chem. Rev. 2010.
We have recently implemented CDFT in the Car-Parrinello molecular dynamics simulation package and applied this method to Ru2+-Ru3+ electron exchange in aqueous solution. Treating solute and solvent at the DFT level of theory and applying the charge constraint on the two Ru hexa-hydrates, the reaction barrier could be computed to good accuracy [JCP09]. Very recently we have extended the code for computation of electronic coupling matrix elements and applied the methodology to study intra-molecular electron transfer in an organic conjugated molecule. Our calculations showed that thermal fluctuations, in particular the slow bending motions of the molecule, lead to variations in the instantaneous electron transfer rate by more than an order of magnitude [JCP10]. Combining free energy and coupling matrix element calculations we recently obtained the absolute reaction rates for Ru2+-Ru3+ electron self-exchange in good agreement with experiment. We showed that electron transfer is most likely at a metal to metal distance of 5.5 Angstroms [Angewandte10]. Current investigations focus on electron transport calculations in organic solar cell materials such as fullerenes (see picture on the left hand side).