We are developing and applying quantum and classical molecular simulation methods to study:
- Charge transport and excited state processes in organic semiconductors
- Electron transfer and electronic conduction in proteins
- Redox processes at solid/liquid interfaces
Have a look at our recent papers:
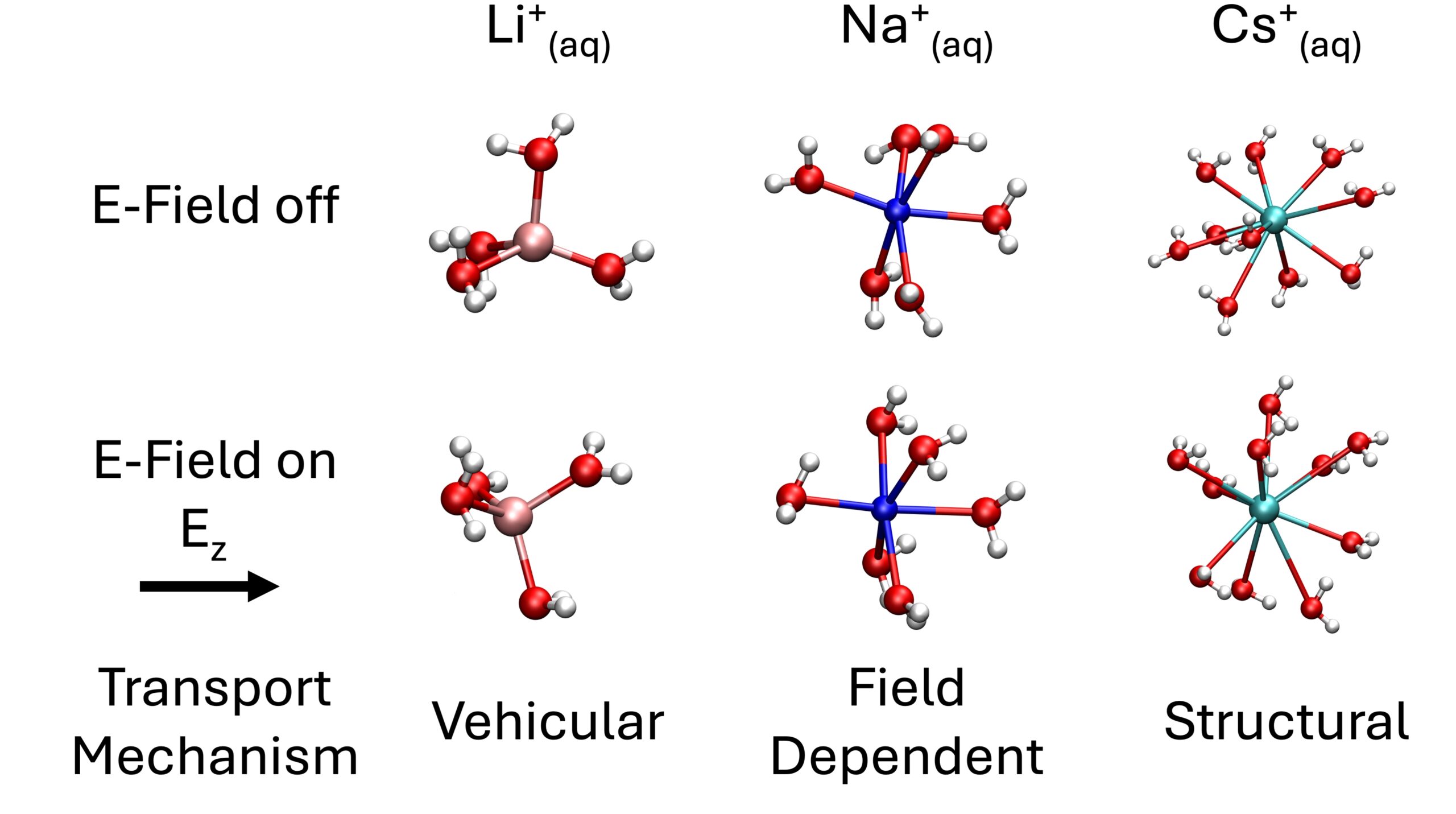
“Transition from Vehicular to Structural Ionic Transport in Electrified Alkali Aqueous Solutions”
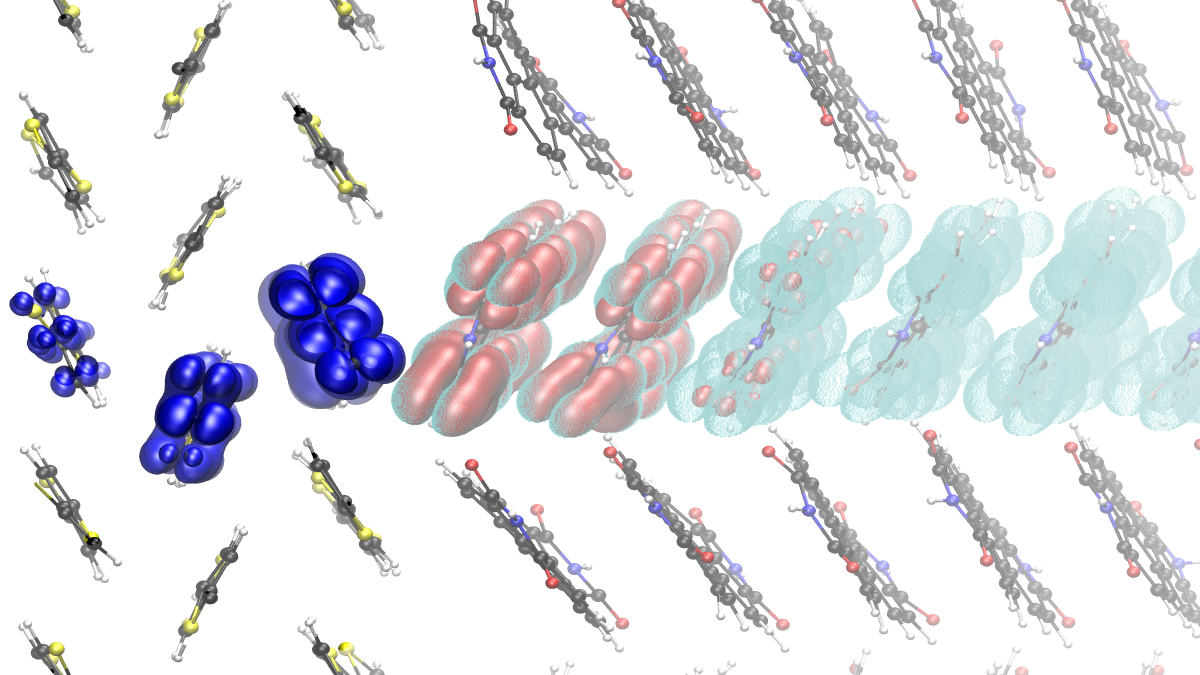
“Transiently delocalised hybrid quantum states are gateways for efficient exciton dissociation at organic donor-acceptor interfaces”
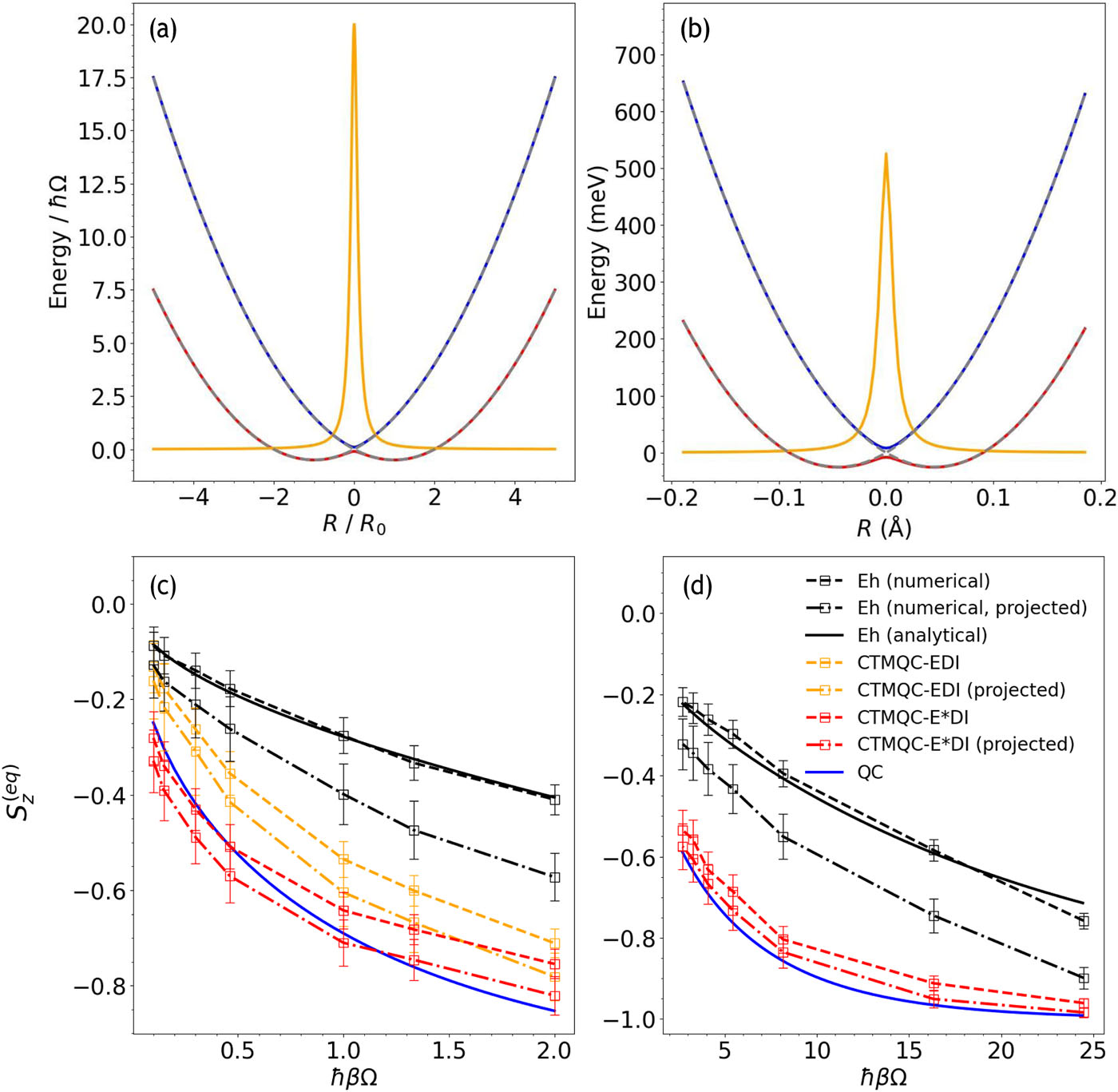
“Thermal equilibrium in coupled trajectory mixed quantum–classical dynamics”
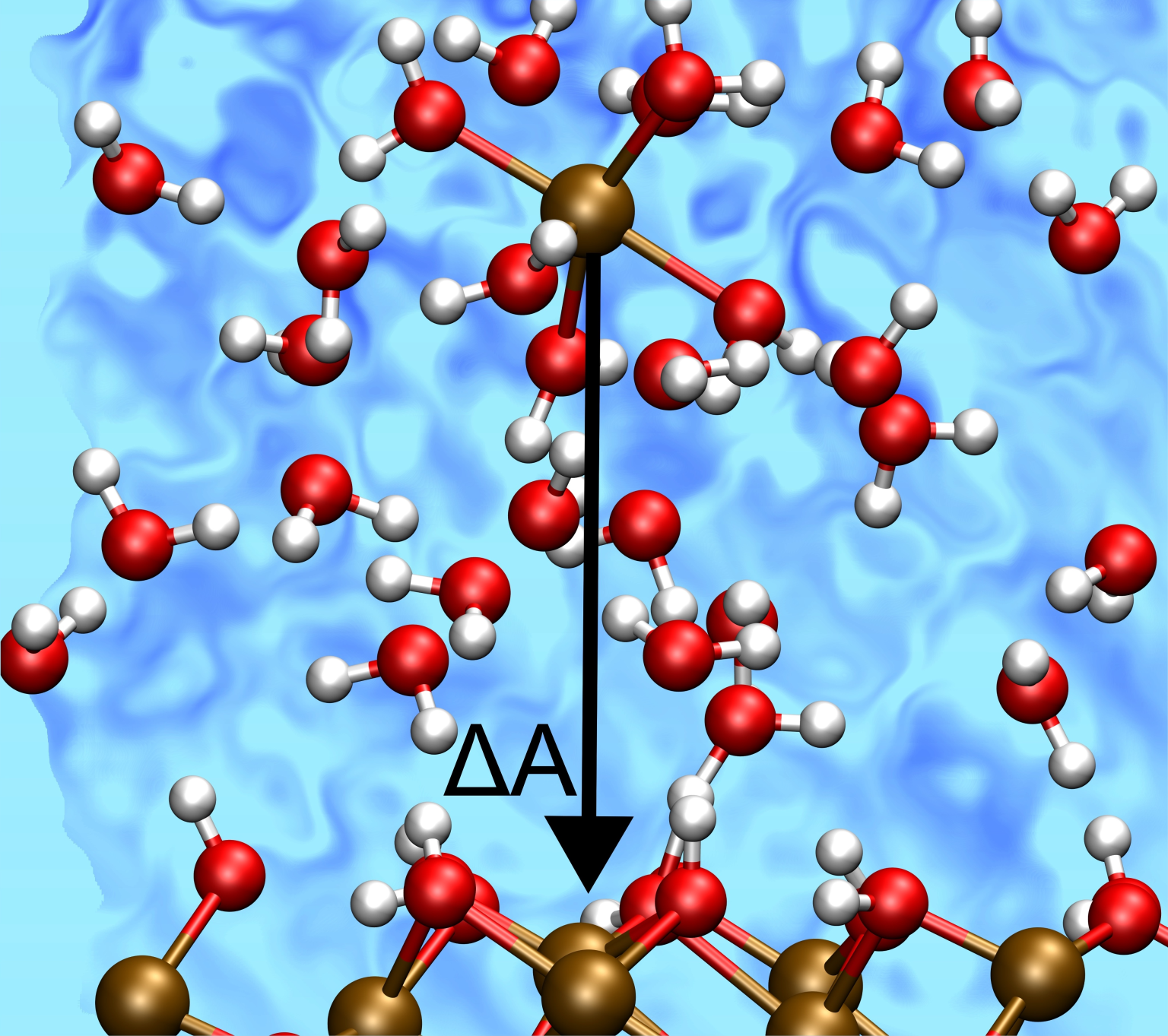
“Mechanism of Fe(II) Chemisorption on Hematite(001) Revealed by Reactive Neural Network Potential Molecular Dynamics”
“Thermoelectric transport in molecular crystals
driven by gradients of thermal electronic disorder”

“Machine learning the electric field response of condensed phase systems using perturbed neural network potentials”

“Disorder-Induced Transition from Transient Quantum Delocalization to Charge Carrier Hopping Conduction in a Nonfullerene Acceptor Material”

“Shallow conductance decay along the heme array of a single tetra-heme protein wire”


Recent News
Jul 26: Andras successfully defended his PhD project on conductance calculations on multi-heme cytochromes - well done Andras!
Jun 26: Group alumnus Philipp Schienbein wins the TYC Early Career Award - congrats Philipp!
Jun 26: Many congratulations to Aaron for winning the best talk prize and Filip for winning the runner-up poster prize at the TYC Student day 2026!
Feb 26: Kit and Filip selected to present their research at the DPG conference in Dresden Germany
Feb 26: Welcome to Jeanne (ENS Paris) who is visiting us for a couple of months to work with Filip on X-SH
Jan 26: Jochen joins the ACS journal J. Chem. Theory Comput. as Associate Editor
Dec 25: Watching excitons dissociate to charge carriers in organic solar cell interfaces - check out Filip's latest non-adiabatic dynamics feat in Nature Communications, https://www.nature.com/articles/s41467-025-67722-4
Oct 25: Andras selected to give a talk at the Young Modellers Forum in Oxford
Sep 25: Welcome to our new PhD student Peter and farewell to Ewen who will spend the next 2 years of his PhD in Singapore!
Jul 25: Ilia and Sayan win prestigious research fellowships - Ilia was awarded a Newton International Fellowship by the Royal Society and Sayan a Walter-Benjamin Fellowship awarded by the DFG - well done, congratulations!!
Jul 25: Filip selected to give a talk at Psi-k 2025 in Lausanne!
Jul 25: Congrats to Andras for the best poster at the Departmental Postgraduate Research Meeting - well done Andras!
Jul 25: Welcome to Sven, who is visiting us from Cologne, and welcome to our new postdoc Ben!
Jun 25: Welcome to our new postdocs Ilia and Sayan!
Jun 25: Kick off to our exciting ERC Advanced Grant EXCITING - special thanks to ERC for selecting it and to UKRI for funding it!
Oct 24: Jan Elsner awarded Marshall Stoneham Prize for best PhD thesis in the Condensed Matter and Materials Physics group (theory/computation) in 2023/24 - congrats Jan!!
Oct 24: Our paper on non-adiabatic dynamics simulation of thermoelectricity is out in Science Advances, exciting work in collaboration with Henning Sirringhaus group, Cambridge - very well done Jan, Aaron and Filip!
Oct 24: Warm welcome to our new PhD students Ewen and Oliver!
Oct 24: Andras to give talk at the MMM Hub conference on his work on multi-heme cytochrome junctions
Sep 24: Our paper on (E-field) Perturbed Neural Network Potentials is accepted in Nature Communications - congrats Kit and Philipp!